Visit our ASCI home page. Below, we show what the inside of the metal Molybdenum (Mo) looks like. The atoms (redish balls) are suspended in smooth electron "sea" (light blue-green).

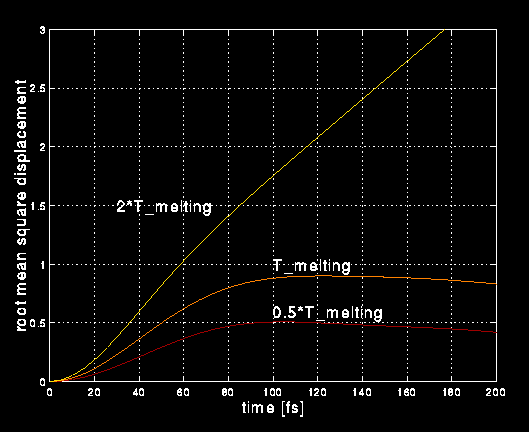
MPEG movies (!) of vibrating nanometer sized bar of silicon, computed using a tight binding model run on Xolas. The algorithm is a fully separable O(N) method which scales well across multiple smp's (less than 15% loss in efficiency). The current calculations show bars consisting of 244 atoms with a cross section of 3 nm X 3 nm in two different microscopic configurations, the so-called "edge" and "zig-zag". Twenty smp's would allow us to run 5000 atoms and allow us to study bars of dimension 15 nm X 15 nm, near the sizes which are accessible experimentally and near where we have predicted a unique size-dependent phase transition. Click on the snap-shot on the left to view the movie for the edge configuration and click on the snap-shot on the right to view the movie for the zig-zag configuration. These results appear to show a more rapid loss of coherence in the edge configutation, suggesting a stronger phonon-phonon coupling for bars in the edge configuration, we plan further studies of this effect.


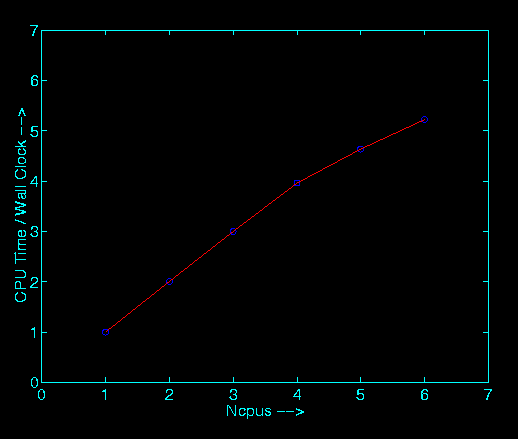
You may also click on the graph below to see the results of more detailed performance analysis.
Code and calculation by Sohrab Ismail-Beigi [Physics].

Code extensions to handle local Stoner parameters and calculation by Dicle Yesilleten [Physics].
Electron density of water (in three dimensions figure on left) and methyline-chloride (in two different two dimensional slices on the right, the first plane containing C-H2 and the second containing C-Cl2) molecules computed using Xolas.


These data then allow us to compute the dipole moment of these molecules, key quantitues in understanding the energetics of Super Critical Water Oxidation (SCWO) reactions, a new promising technology for breaking down toxic wastes.
The table below summarizes our results:
| Dipole moments (Debye units) | This work (Debye units) | Experiment (Debye units) | Error |
|---|---|---|---|
| H2O | 1.85 | 1.89 | -2% |
| Methyline chloride (C-H2-Cl2) | 1.78 | ~1.6 | +11% |
| H2O-C-H2-Cl2 activated complex | Unknown | ||
For more techical details about SCWO and the relevance of this calculation, click here.
Xolas implementation of code by Parry Husbands [EECS, supervised by Prof. Alan Edelman], calculation by Phil Marrone and Jason Cline [ChemE].
Here we show the electron density in two different planes sliced through a crystal of Ge, calculated using ab initio quantum mechanical methods on Xolas with two different algorithms.
The first algorithm is a traditional plane wave calculation where the 3D FFT is coded using threads running in parallel on a single smp. The results for this traditional approach appear below in red.
The second algorithm uses a different representation of quantum mechanics, the density matrix approach. The code, still under development, currently runs in MPI distributed across three of the smp's with a negligible loss in efficiency (again less than 15%). Preliminary results, computed at lower resolution, are pictured in green for comparison. While the current calculations contain only 64 atoms, with 20 smp's, we anticipate being able to perform calculations in systems with approximately 1000 atoms. These will represent the largest electronic structure calculations ever carried out and will allow us to gain deep insights into the mechanical behavior of materials.

 (100 plane)
(100 plane)

 (110 plane, showing bonds)
(110 plane, showing bonds)
Xolas implementation of traditional code by Parry Husbands [EECS, supervised by Prof. Alan Edelman], code for new method developed by Sohrab Ismail-Beigi [Physics]; calculations by Sohrab Ismail-Beigi [Physics].
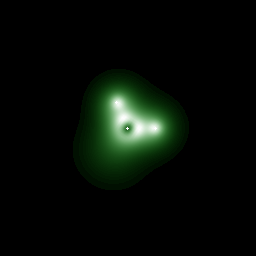
Calculation of a key process, known as antiphase defect (APD) migration, which controls the mechanical behavior of silicon. This calculation was run on a single smp, involves 144 atoms and is at the limit of what a single smp can do. To make significant progress in the understanding of the mechanical properties of silicon will require calculations with on the order of 1000 atoms, which are foreseeable on a cluster of 20 smp's. The figure below shows the density of electrons at the transition state for migration. The atom in the center of the first figure is about to move from one partner to another. The second figure illustrates the context of the migration process, in which the bonded shaded atoms switch partners.
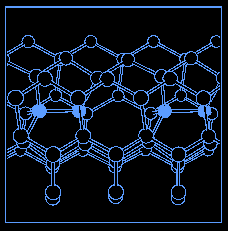
Xolas implementation of code by Parry Husbands [EECS], calculations by Gabor Csanyi [Physics].
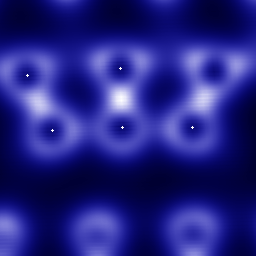
The previous study focuses on the electronic aspects of APD migration. We also use Xolas to study the atomistic configurations related to this reaction. Here we use Monte Carlo studies, which ideally suited to loosely coupled smp's, to explore the phase space of the reactions. The figure below shows the thermal occupation of the reaction pathway as the APD migrates. The first figure shows the pathway relative to the surrounding lattice. (The blue balls represent the positions of the atoms before the reaction; the red sticks with arrows indicate the covalent bonds; the green and beige regions show the probability density for the position of one key atom during the reaction which breaks the bond that runs approximately vertically in the figure.)
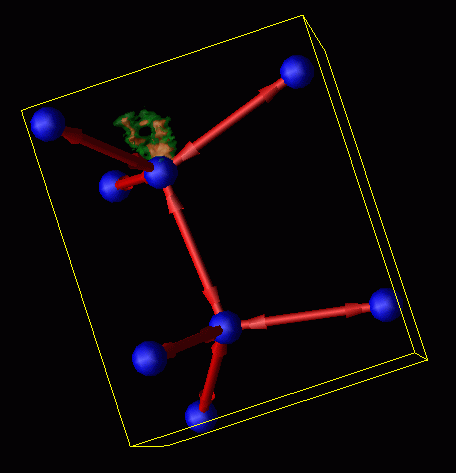
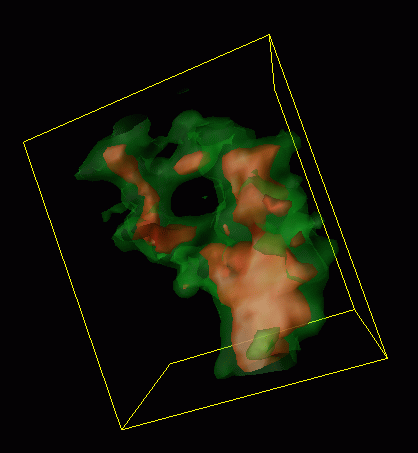
The second figure shows a blow-up of the thermal probabilistic pathway. The "hole" in the middle of this distribution indicates that a previously unknown pathway is at work in this process. This discovery, made with the Xolas project, has changed our view of the reaction mechanism at its implications are currently under investigation.
More computations are required to improve the statistics of these studies to where they will be useful. We require a five-fold improvement in our statistics (to reduce the typical errors from 0.1 eV down to room temperature, ~0.02 eV), which requires twenty-five times the sampling. A cluster of twenty smp's would be ideal for this study. Such improvement in sampling will also allow us to manage artificial correlations present in the studies we were able to carry out with a single smp.
Code and calculations by Torkel Engeness [Physics].